近日,数学与交叉科学研究中心在冷冻电镜三维结构比对上取得新进展,相关研究成果在nature communications以及genomics, proteomics & bioinformatics期刊上发表。
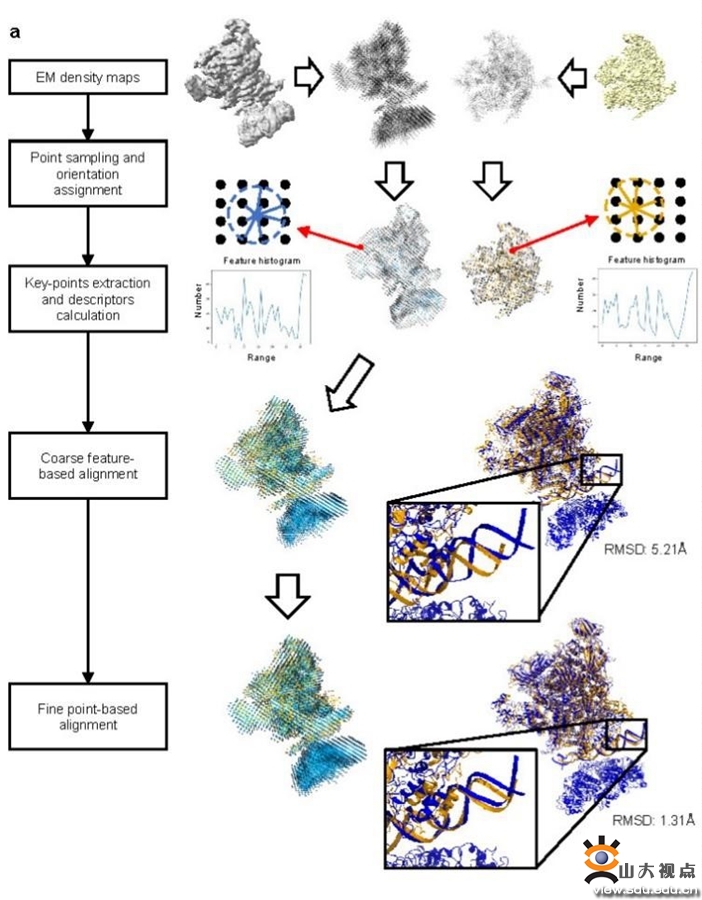
图1 cryoalign比对流程示意图
中心博士研究生何彬涛在nature communications发表了题为“accurate global and local 3d alignment of cryo-em density maps using local spatial structural features”的研究论文。该论文提出了一种基于局部结构特征的冷冻电镜密度图三维体对齐方法cryoalign。cryoalign充分探究了密度图本身密度值分布特点,利用点云表示替代冗余的体素信息,设计基于局部密度值变化趋势的空间结构特征表示,构建两阶段比对策略,从特征域到空域逐步达到高效高精度的结构比对结果。实验结果表明,cryoalign相较于现有方法表现更优,并在后续的密度图比较以及原子模型组装流程中发挥有效作用。山东大学数学与交叉科学研究中心韩仁敏、沙特阿卜杜拉国王科技大学高欣为通讯作者,北京理工大学张法团队为合作方。
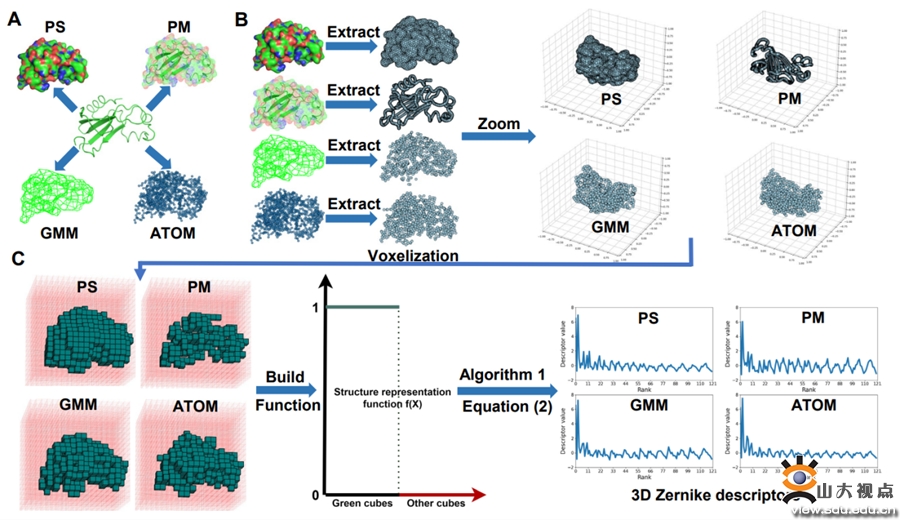
图2 fp-zernike算法的工作流程
同时,中心博士研究生祁俊海在genomics, proteomics & bioinformatics发表了题为“fp-zernike: an open-source structural database construction toolkit for fast structure retrieval”的研究论文。该工作主要针对蛋白质模型数据库的爆炸式增长的情况下,如何进行快速结构检索的问题。特别的alphafold2的发布引发了蛋白质模型数据库的爆炸式增长,这为研究生物结构的功能提供了更多资源。结构检索是分析结构模型的关键手段,而结构检索的核心是通过结构比对来衡量结构之间的相似性。尽管现有的结构比对算法可以解决这一挑战,但它们通常耗时。为了克服这一限制,开发了fp-zernike工具包,它基于特征点来计算不同类型的zernike描述符,从而将蛋白质(rna)结构用固定长度的向量来表示,同时将结构检索的问题转换成欧式距离的计算问题,这大大降低了检索问题的复杂度。基于fp-zernike来计算不同类型的zernike描述符的主要有以下步骤:(1)获取表面、网格和原子点云的结构表示;(2)提取并缩放特征点至单位球面;(3)基于特征点构建描述结构的函数;(4)计算相应的几何矩;以及(5)使用特定的数学表达式计算3d zernike描述符。这五个步骤在下图中简要描述。在多个数据集上对fp-zernike进行了测试。结果表明,相对于当前主流的结构检索工具,fp-zernike能更准确、更快速的完成结构比对。特别地,fp-zernike表现出非常明显地效率优势,比其他结构比对工具快了接近1000倍。这是因为fp-zernike通过计算结构描述符之间的欧氏距离来衡量结构相似性,其固定的描述符长度导致时间复杂度为o(1)。山东大学数学与交叉科学研究中心李国君、韩仁敏为论文通讯作者。


 微信公众号
微信公众号
